Hemochromatose Vereniging Nederland (HVN) (Nederlands)
In the case of haemochromatosis, the intestines absorb more iron from processed food than is required. Normally this excess is removed through urine, but in the case of patients with hemochromatosis, the concentration of iron is too high to be normally processed. As a result, iron accumulates in different organs around the body. Also known as iron overload disorder, haemochromatosis is generally a hereditary condition.
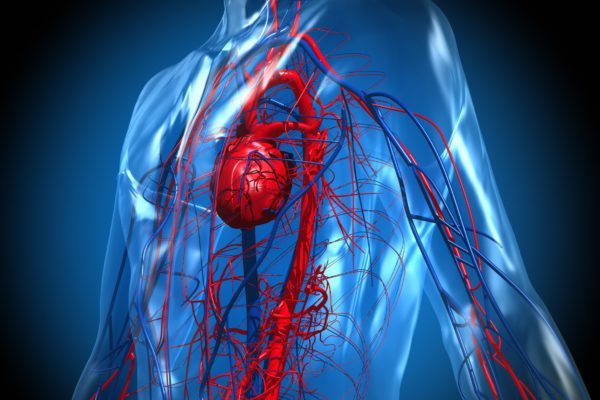
In the early stages of the disease, the iron overload does not have serious consequences and is stored primarily in the liver. However, as the concentration of iron increases, the liver struggles to store it all, resulting in liver damage by an increase of connective tissue. Eventually this leads to the development of liver cirrhosis. After the liver is damaged, the iron will accumulate in other organs, such as the pancreas, the muscular walls of the heart, the skin and joints around the body.
The symptoms of haemochromatosis are not the same for every person. It presents itself mostly in vague complaints such as chronic fatigue, joint problems, abdominal complaints, diabetes, liver function abnormalities, hormonal disorders, skin discoloration and loss of libido. Due to a large variety of symptoms, a physician may not immediately consider haemochromatosis. Furthermore, most patients do not have clear symptoms of the disease in later stages of life, and some patients may not even have symptoms, despite having haemochromatosis.
Furthermore, iron accumulation in organs can cause specific complaints:
There are two types of haemochromatosis: primary and secondary haemochromatosis.
Because haemochromatosis presents itself with a wide variety of symptoms, it may take a while to diagnose the disease. The final diagnosis is based on the following studies:
The treatment of hereditary or primary haemochromatosis is aimed at preventing further liver damage. The treatment consists of:
In patients with the secondary type of hemochromatosis, iron chelation therapy is the most common provided therapy. Iron-binding agents (deferasirox, deferiprone, deferoxamine) can also be prescribed to achieve normal iron levels.